

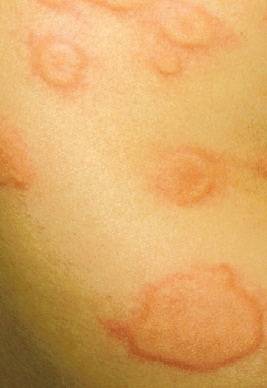
El angioedema hereditario es una afectación genética poco frecuente que se manifiesta en 1:10,000 -1:50,000 individuos.
Se origina por un defecto genético en el cromosoma 11 que provoca una deficiencia o mal funcionamiento de una proteína llamada inhibidor de C1-esterasa, que forma parte de un grupo de moléculas conocidas como Sistema de Complemento.
Esta proteína controla, entre otras cosas, la síntesis y liberación de la bradicinina, que es un potente mediador de la inflamación.
Este defecto trae como consecuencia que se produzcan episodios graves de hinchazón de 2-5 días de duración, que puede afectar tanto la piel del cuerpo como la cara, cuello, labios, párpados, extremidades, así como toda la mucosa respiratoria y gastrointestinal.
Los episodios pueden llegar a provocar dificultad respiratoria e incapacidad para tragar, que si no se tratan pueden llevar a la muerte.
La inflamación del intestino puede provocar cuadros intensos de dolor abdominal, que a menudo se confunden con apendicitis.
Los síntomas suelen aparecer en la infancia, los episodios ocurren de forma espontánea y ceden en unas 6 horas.
Al ser una enfermedad de origen genético, suele haber antecedentes en la familia, salvo en casos de mutación de Novo (es decir, cuando se presenta por primera vez en la familia).
Las crisis de angioedema hereditario pueden ser desencadenadas por distintas circunstancias, como traumatismos, intervenciones quirúrgicas, extracciones dentarias, mal descanso nocturno y estrés.
El angioedema hereditario puede empeorar por los factores hormonales, como los estrógenos, toma de anticonceptivos orales y tratamientos hormonales sustitutivos.






